On December 12, 2019, Qing Zhou's group from the Life Sciences Institute of Zhejiang University, Junying Yuan 's group from Harvard Medical School, and Xiaochuan Wang 's group from Children’s Hospital of Fudan University jointly published a research article in Nature entitled A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1, which reported that the RIPK1 cleavage site variants caused severe auto-inflammatory diseases. The revealed molecular mechanism of the disease guided clinical treatment and greatly relieved the clinical symptoms of patients.

Activation of RIPK1 controls TNF-mediated apoptosis, necroptosis and inflammatory pathways. Cleavage of human and mouse RIPK1 after residues D324 and D325, respectively, by caspase-8 separates the RIPK1 kinase domain from the intermediate and death domains. The D325A mutation in mouse RIPK1 leads to embryonic lethality during mouse development. However, the functional importance of blocking caspase-8-mediated cleavage of RIPK1 on RIPK1 activation in humans is unknown. Here we identify two families with variants in RIPK1 (D324V and D324H) that lead to distinct symptoms of recurrent fevers and lymphadenopathy in an autosomal dominant manner. Impaired cleavage of RIPK1 D324 variants by caspase-8 sensitized patients’ peripheral blood mononuclear cells to RIPK1 activation, apoptosis and necroptosis induced by TNF. The patients showed strong RIPK1-dependent activation of inflammatory signalling pathways and overproduction of inflammatory cytokines and chemokines compared with unaffected controls. Furthermore, we show that expression of the RIPK1 mutants D325V or D325H in mouse embryonic fibroblasts confers not only increased sensitivity to RIPK1 activation-mediated apoptosis and necroptosis, but also induction of pro-inflammatory cytokines such as IL-6 and TNF. By contrast, patient-derived fibroblasts showed reduced expression of RIPK1 and downregulated production of reactive oxygen species, resulting in resistance to necroptosis and ferroptosis. Together, these data suggest that human non-cleavable RIPK1 variants promote activation of RIPK1, and lead to an autoinflammatory disease characterized by hypersensitivity to apoptosis and necroptosis and increased inflammatory response in peripheral blood mononuclear cells, as well as a compensatory mechanism to protect against several pro-death stimuli in fibroblasts.
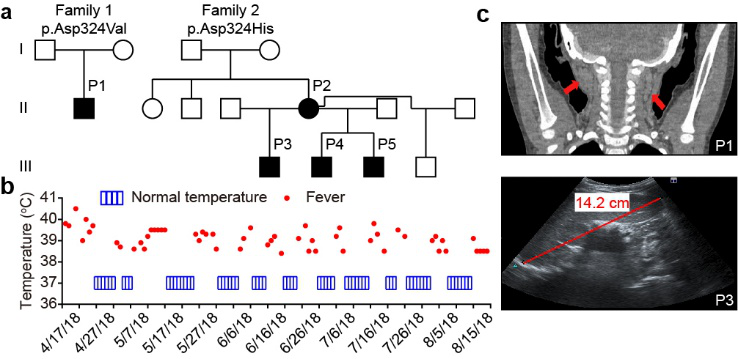
Fig. 1 Heterozygous variants at the RIPK1 cleavage site cause autoinflammatory disease.
Ph.D. candidate Panfeng Tao and Jun Wang and Dr. Shihao Wang of Qing Zhou's group are co-first authors. This research was supported by National Key Research and Development Project, The National Natural Science Foundation of China, Zhejiang Provincial Natural Science Foundation of China and the Fundamental Research Funds for the Central Universities.
Text link:https://www.nature.com/articles/s41586-019-1830-y