On March 19th, 2024, Dong Fang’s research team published an online research paper entitled FUS reads histone H3K36me3 to regulate alternative polyadenylation in Nucleic Acids Research, reporting that FUS as an H3K36me3 reader protein regulates APA in cells.
Recently, chromatin structure and epigenetic histone modifications emerged as novel regulators of nascent RNA maturation. The cleavage and polyadenylation in the 3 ' termini are important for translation and mRNA stability1. For many eukaryotic transcripts, alternative polyadenylation (APA) generated multiple transcript isoforms with diverse 3'UTR which recruit different cis-regulatory elements2. Dysregulation of APA is frequently found in many human diseases, including cancer and neurodegeneration3. The trimethylation of histone H3 at lysine 36 (H3K36me3) is enriched at the 3 ' end of gene bodies, particularly those of highly expressed genes4. Therefore, this study proposed that H3K36me3 may participate in the regulation of polyadenylation at the 3'UTR, related to poly(A) length or APA sites.
FUS has been involved in multiple levels of RNA processing including transcription, splicing, transport and so on5. Mutations in the Fus gene cause amyotrophic lateral sclerosis (ALS)6, a neuron degenerative disease. Here, this study identified FUS as a reader for H3K36me3. Depletion of H3K36me3 at gene bodies released FUS from chromatin and increased the interaction of FUS with mRNA, which, in turn, led to APA selection being distant from the stop codon. The binding between FUS and H3K36me3 depends on proline residues in the ZNF domain of FUS, and therefore, mutations of proline residues cause loss of chromatin association and increased mRNA binding, resulting in an increase in distal APA selection. Moreover, loss of H3K36me3 recognition by FUS, which is mimicked by a proline mutation in FUS in ALS, contributes to hyperactivation of mitochondria and hyperdifferentiation in mouse embryonic stem cells (mESCs) (Figure 1).
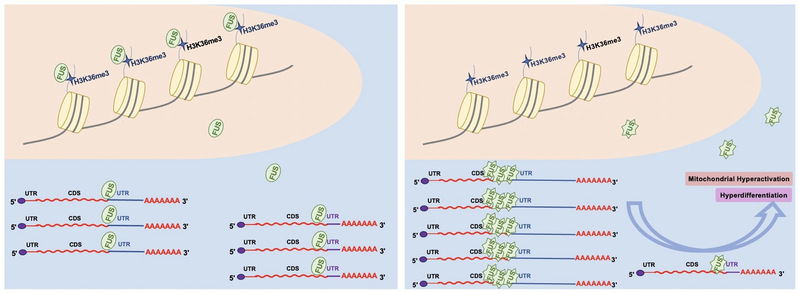
Figure 1. A model showing the function of H3K36me3 recruited FUS in APA selection.
In this study, the authors reconstituted H3Kc36me3 nucleosomes which mimicked the tri-methylated H3K36 for pulldown assay in vitro and designed Biotinylated Oligo coupled polyadenylation profiling With Long-read sequencing (Bowl-seq) to identify poly(A) changes. To reveal how H3K36me3-regulated APA affects cellular function, the authors test the reactive oxygen species (ROS) and the oxygen consumption rate (OCR), and applied embryoid body (EB) formation assay. Together, these observations led to the suggestion that FUS is an H3K36me3 reader protein that links chromatin-mediated alternative polyadenylation to human disease. These data expand our understanding of how H3K36me3-regulated APA affects mitochondrial function and cell differentiation and shed light on the causative effect of FUS mutations in ALS.
Dong Fang is the corresponding author of this article. Jun qi Jia, Haonan Fan, and Xinyi Wan, from Life Sciences Institute, Zhejiang University, are the co-first authors. This work is supported by Professor Jun Huang. This work is also supported by the National Key R&D Program of China, the National Natural Science Foundation of China, and the Zhejiang Provincial Natural Science Foundation of China.
References
1. Tian, B. & Graber, J.H. Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip Rev RNA (2012).
2. Tian B, Manley J L. Alternative polyadenylation of mRNA precursors. Nature Reviews Molecular Cell Biology (2016).
3. Mitschka, S. & Mayr, C. Context-specific regulation and function of mRNA
alternative polyadenylation. Nat Rev Mol Cell Biol (2022).
4. Leung, C.S. et al. H3K36 Methylation and the Chromodomain Protein Eaf3 Are Required for Proper Cotranscriptional Spliceosome Assembly. Cell Rep (2019).
5. Yu, Y. & Reed, R. FUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNP. Proc Natl Acad Sci U S A (2015).
6. Kwiatkowski, T.J., Jr. et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (2009).
Link to the paper:https://doi.org/10.1093/nar/gkae184