2017年4月26日,张龙实验室在Nature Communications发表题为“FAF1 phosphorylation by AKT accumulates TGF-β type II receptor and drives breast cancer metastasis”的文章,该文章阐释了发挥肿瘤抑制功能的TGF-β信号是如何被扭转为促进肿瘤转移的驱动力的这一亟待回答的问题。
肿瘤转移是导致癌症病人死亡的一个非常重要的原因,多达90%的实体瘤患者死于肿瘤转移。转移生长因子β (TGF-β)信号通路在肿瘤发生和发展中有着非常重要的作用。TGF-β在早期癌变中抑制肿瘤生长,而在后期肿瘤发展过程中则转变成一个肿瘤促进因子。TGF-β在肿瘤发展后期如何转变成为促癌因子? 这些转变是否与其他原癌信号的影响有关等都是研究肿瘤转移的关键问题,这些问题的解答将为后期肿瘤转移的治疗和药物研发提供理论基础。
作为TGF-β 配体结合的第一个分子,TGF-β的II型受体(TβRII)除了激活经典的SMAD通路,还可以通过磷酸化或与信号中间体结合激活包括磷脂酰肌醇激酶(PI3K)/AKT等在内的非经典SMAD信号通路。在乳腺癌晚期,高水平TGF-β诱导的侵润性和转移性和过度激活的PI3K/AKT信号密切相关。然而两者之间的分子机制并不清楚。目前对细胞膜上TβRII代谢的机制研究得较少,特别是TβRII蛋白水平的变化对肿瘤发生发展的影响也没有一致的观点,暗示其在早期和晚期肿瘤中的不同作用或者其他原癌信号对其功能产生了影响。
本文章从研究受体的结合蛋白入手,发现TβRII受到FAF1这个接头蛋白的严密控制。FAF1通过招募VCP和E3连接酶复合物限制了细胞膜上TβRII的水平。由于极低的TβRII就能保证持续不断地磷酸化TβRI从而发挥SMAD介导的正常功能,这种调节方式既保持了下游SMAD信号的正常传递,又防止了TβRII的过度激活。而在肿瘤发展后期,尤其是当肿瘤响应刺激或者接受诱导获得AKT的激活后,FAF1的特异性丝氨酸位点会被AKT磷酸化从而脱离细胞膜并丧失上述功能,导致膜上TβRII的水平上调,激活SMAD和非经典SMAD信号通路,促进肿瘤转移。另一方面,AKT又能阻断SMAD介导的细胞周期调控,因而TGF-β/SMAD能快速地被扭转成为促进肿瘤转移的驱动力。
此项研究不仅发现了调控细胞膜上TβRII蛋白代谢的分子机制,而且解释了在恶性肿瘤细胞中AKT如何通过FAF1逆转TGF-β信号促进肿瘤转移的原因,对临床预防肿瘤恶化和抗癌药物的研究提供了重要的理论支持。
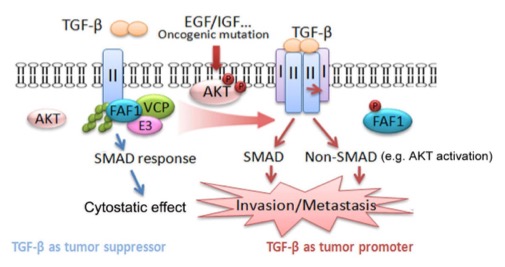
图:FAF1的磷酸化失活促进乳腺癌的转移。
本文的第一作者是博士生谢枫,金珂和邵丽,张龙教授为本文的通讯作者。