2019年12月12日,浙江大学生命科学研究院周青课题组与哈佛大学医学院袁钧英课题组、复旦大学附属儿童医院王晓川课题组联合在《Nature》杂志上发表研究论文“A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1”,报道了RIPK1变异导致新的自身炎症性疾病,揭示了致病分子机制,指导临床治疗缓解了病人的临床症状。
背景介绍
自身炎症性疾病(autoinflammatory disease)的主要临床表现为周期性发热,同时可伴有皮疹、淋巴结肿大、关节炎/关节痛、口腔溃疡、血管炎/血管病变、腹痛/腹泻、间质性肺病、脑基底节钙化等其中一部分症状的一类疾病,而且炎症标志物 C反应蛋白升高。很多患有自身炎症性疾病的病人由于没有认识到这是一类遗传病,得不到遗传确诊,辗转全国各医院的风湿免疫科,神经内科,呼吸科、消化科、皮肤科,却得不到有效的治疗。
RIPK1 是受体相互作用蛋白(RIP)激酶家族的一员,参与了决定细胞“生死存亡”的多种重要信号通路,其激酶活性在RIPK1依赖的细胞凋亡和细胞程序性坏死进程中发挥重要的调节作用。RIPK1蛋白的中间结构域中含有Caspase-8的切割位点(Asp324)(图1)。以往的研究发现,小鼠RIPK1该位点突变会导致胚胎致死,主要是由于增多的细胞凋亡和程序性坏死。然而,人类RIPK1该切割位点变异对这些重要信号通路及人类健康的影响未有报道。
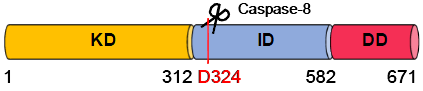
图1 RIPK1蛋白模式图
文章解读
周青课题组通过全外显子测序鉴定到了两个分别携带有D324不同位点新发突变的家系,病人表现出典型的自身炎症疾病表型,主要包括周期性发热、淋巴结肿大、肝脾肿大等(图2),病人的血清和外周血单核细胞(PBMC)均表达较高水平的炎症因子(如IL-6、TNF等)。
| |
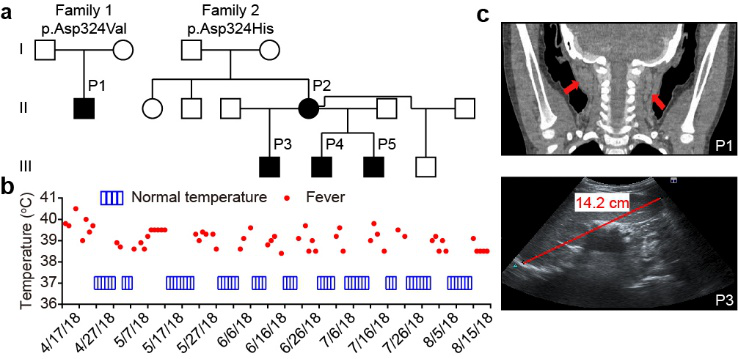
图2 病人家系信息及临床表型
在病人PBMC细胞和小鼠MEFs细胞中研究发现,RIPK1突变细胞对TNF诱导的细胞凋亡和细胞程序性坏死更加敏感,并且可以诱导产生显著上调的IL-6、TNF等炎症因子。这些变化都依赖于RIPK1的激酶活性,可以通过RIPK1激酶抑制剂Nec-1s缓解。有趣的是,与病人PBMC相反,病人的皮肤成纤维细胞表现出对TNF诱导的细胞死亡抵抗,而且对其他刺激如LPS、TRAIL诱导的细胞死亡以及Erastin、RSL3等诱导的铁死亡也有抵抗作用。进一步研究发现,病人成纤维细胞RIPK1、TNFR1等蛋白表达水平明显下调,细胞内的还原性谷胱甘肽(GSH)含量高,活性氧(ROS)含量低,这些变化部分解释了成纤维细胞对不同死亡形式的抵抗。这一现象提示病人成纤维细胞为应对RIPK1变异导致的对多种刺激的高敏感性发展出多种补偿机制来维持机体稳态。
本研究首次发现人类RIPK1非切割变异导致一种新的自身炎症性疾病。解析其发病分子机制,不能切割的RIPK1可以促进其激酶活性,导致细胞凋亡、细胞程序性坏死增加,引起炎症因子上调;同时还发现病人不同种类细胞响应外来刺激的异质性,丰富了RIPK1在调节不同种类细胞死亡中的作用。并对病人进行了anti IL6的治疗,使病情得到有效的改善。
周青课题组博士生陶攀峰、王俊和博士王诗豪为本文共同第一作者,周青研究员、袁钧英教授、王晓川教授和俞晓敏博士为共同通讯作者。该研究获得了国家重点研发计划项目、重大项目自然科学基金委面上项目、浙江省自然科学基金委杰出青年科学基金的资助。
原文链接:https://www.nature.com/articles/s41586-019-1830-y