2026年3月16日,我院蒋超实验室在国际著名生物信息学期刊Briefings in Bioinformatics发表研究成果,推出新一代宏基因组分类软件鲲鹏(Kun-peng)。其名取自著名中国神话生物,寓意兼具宏阔视野与迅捷之势。面向跨域宏基因组分析中长期存在的高内存、高成本和高门槛难题,鲲鹏以极低内存、高速度、高灵敏度和低假阳性的综合优势,实现了性能与可及性的同步提升,为复杂环境样本和超大参考数据库的高效解析提供了新的技术路径,也推动超大规模pan-domain宏基因组分析从稀缺高性能平台走向个人电脑和更广泛实验室场景中的实际应用。
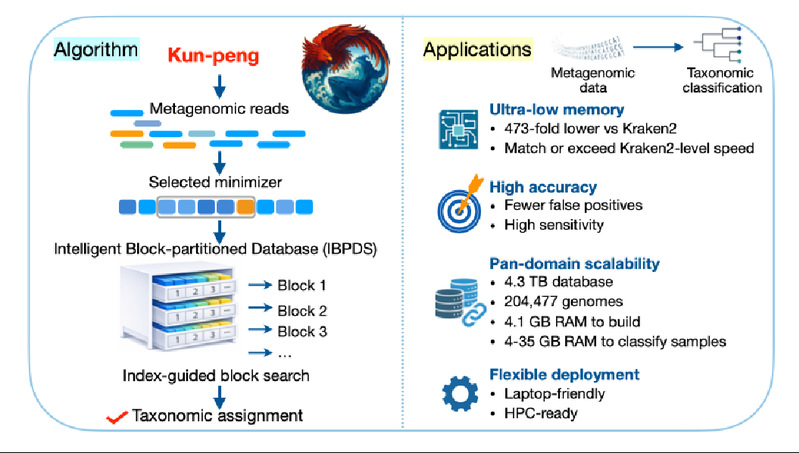
宏基因组技术已成为解析复杂微生物群落的重要手段,但随着参考基因组数量持续增长,尤其是在细菌、古菌、病毒、真菌、植物和动物等多个生物域共同纳入分析框架后,传统方法越来越受到计算资源的限制。数据库越大,往往意味着越高的内存需求和越强的硬件依赖,这在很大程度上限制了跨域宏基因组分析的推广,也使超大规模参考数据库的价值难以在常规科研条件下得到充分发挥。
针对这一关键瓶颈,鲲鹏围绕数据库组织方式与分类搜索流程进行了系统优化,建立了一套适用于超大规模参考数据库的高效分类方法。研究显示,鲲鹏可仅用 4.1 GB 峰值内存构建 4.3 TB 的超大pan-domain数据库;在样本分类阶段,速度最高可达 Kraken2 的 4.73 倍,分类内存占用则可降低 54–473 倍。这意味着,以往依赖高内存服务器完成的跨域宏基因组分析,如今在个人计算机上也具备了现实可行性。对于许多实验室而言,这不仅是性能上的优化,更意味着超大规模跨域分析门槛的实质性下降。
同时,鲲鹏在分类质量控制方面也展现出突出优势。相较 Kraken2、KrakenUniq 和 Centrifuger 等多款代表性工具,鲲鹏在多组数据中的假阳性水平更低,部分场景下甚至优于 mOTUs。对于复杂样本分析而言,较低的假阳性意味着更高的结果可信度,也意味着研究者能够在更大参考数据库中获得更稳健、更可靠的分类结果。这表明,鲲鹏的优势不仅体现在计算效率和资源友好性上,也体现在分类结果的稳定性与可信度上。
除性能与分类质量优势外,鲲鹏还具备良好的兼容性与应用延展性。该软件可无缝衔接 Kraken2 数据库体系,便于研究者在现有数据库基础上直接开展分析;同时兼容后续 Bracken 的丰度估算流程,有助于在不显著改变既有分析管线的前提下,实现从序列分类到丰度计算的顺畅衔接。这一特性显著降低了工具迁移与应用部署的成本,有利于鲲鹏在现有宏基因组分析体系中的快速推广。
在真实复杂样本分析中,鲲鹏的价值进一步凸显。面对空气、水体、土壤和人体相关等来源复杂、背景多样的宏基因组样本,marker-based 方法往往由于灵敏度较低,难以充分识别复杂样本中的广泛生物组成;而以 Kraken2 为代表的主流 k-mer 方法,虽然具有较强的分类能力,但在超大 pan-domain 数据库条件下又常受制于较高的内存消耗和运行负担,难以在普通硬件环境中真正发挥大数据库的优势。相比之下,鲲鹏同时兼具大数据库承载能力、复杂样本高灵敏度和资源友好性,能够显著提升 reads 的分类覆盖度,使超大规模 pan-domain 数据库不再停留于理论层面的可构建,而真正具备了在实际科研场景中广泛应用的可行性。这也使鲲鹏在复杂环境样本和跨域生物组研究中展现出独特而重要的应用价值。
鲲鹏的发布,为 pan-domain 宏基因组分类提供了一种兼具高性能与高可及性的解决方案。正如鲲鹏之名所寓意的那样,它不仅实现了对超大参考数据库的高效驾驭,也打通了跨域宏基因组分析从高算力依赖走向广泛应用的关键一步。随着公共参考基因组资源持续扩展,鲲鹏有望进一步推动宏基因组研究朝着高效化、普及化和跨域化方向发展。
陈琼博士和张博良为论文的共同第一作者,蒋超研究员为论文的通讯作者。参与该研究的还有蒋超实验室的博士生彭晨和刘桢博士,以及新加坡南洋理工大学的申小涛教授。研究课题受国家自然科学基金项目资助。
原文链接:https://doi.org/10.1093/bib/bbag119