

1. Smad4 sequestered in SFPQ condensates prevents TGF-β tumor-suppressive signaling
(Developmental Cell. 2023 Dec 15)
TGF-β是调控细胞稳态的多功能细胞因子,它在肿瘤发生早期通过诱导细胞周期阻滞和促进细胞凋亡发挥抑癌作用。作为TGF-β信号转导的中心蛋白Smad4,其活性的丧失是肿瘤发生的关键事件,在胰腺和大肠癌中常见Smad4基因的缺失和突变。然而,非突变性Smad4活性丧失的肿瘤特异性机制尚不清楚。本研究发现一种叫做SFPQ的RNA结合蛋白直接结合Smad4,通过相分离抑制了Smad4的转录活性,从而阻断了TGF-β信号的抑瘤功能。
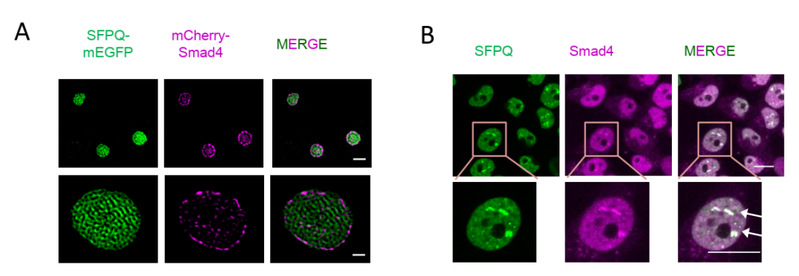
图 1 SFPQ凝聚体在分子(A)和细胞(B)水平滞留抑癌蛋白Smad4
相分离现象是近年来的热点方向,细胞内大分子可以通过相分离形成无膜结构的凝聚体,在细胞信号传导、物质输运、免疫应答等细胞功能方面扮演了重要角色。相分离不仅可以浓缩聚集细胞内的特定大分子从而加速生物化学反应,还可以在时间和空间上隔断不同的成分从而负调控反应进程。过去的工作发现SFPQ能形成RNA结合依赖的相分离。本研究发现,SFPQ对TGF-β信号的调控并不依赖于它的RNA结合能力,而是通过类朊病毒结构域(PrLD)形成液-液相分离,并通过新型的Smad结合结构域,在细胞核内将Smad4选择性地滞留在凝聚体内,导致Smad复合物的解离,破坏了Smad4的转录活性。进一步的临床数据分析显示SFPQ的高表达与多种人类癌症(如早期原发性肝细胞癌)的发生密切相关。不同的肝癌模型证实了SFPQ能够促进肿瘤形成,其促癌作用依赖于相分离活性。因此,本研究揭示了相分离在细胞信号转导的时空调控作用,提出了肿瘤细胞拮抗TGF-β生长阻滞作用的新机制,为癌症的早期诊断和靶向治疗提供新的思路。
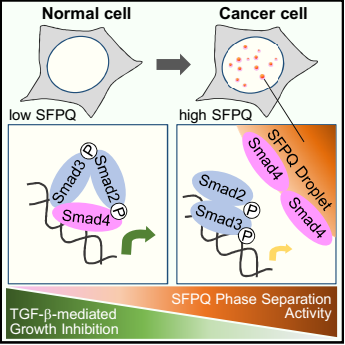
图 2 SFPQ凝聚体阻断TGF-β抑制肿瘤发生的分子机制工作模型
2.Imatinib blocks tyrosine phosphorylation of Smad4 and restores TGF-β growth-suppressive signaling in BCR-ABL1-positive leukemia.
(Signal Transduct Target Ther. 2023 Mar 24; 8(1): 120.)
TGF-β介导的生长抑制的丧失是癌症发展的主要原因,最典型的例子是结直肠癌和胰腺癌中编码TGF-β信号通路成分的基因的功能缺失突变。或者,功能获得癌基因突变也会破坏抗增殖TGF-β信号传导。然而,致癌基因诱导的TGF-β信号传导调控的分子机制尚未得到广泛研究。本研究发现慢性粒细胞白血病(CML)的致癌BCR-ABL1和细胞ABL1酪氨酸激酶磷酸化并失活Smad4,以阻断抗增殖TGF-β信号传导。
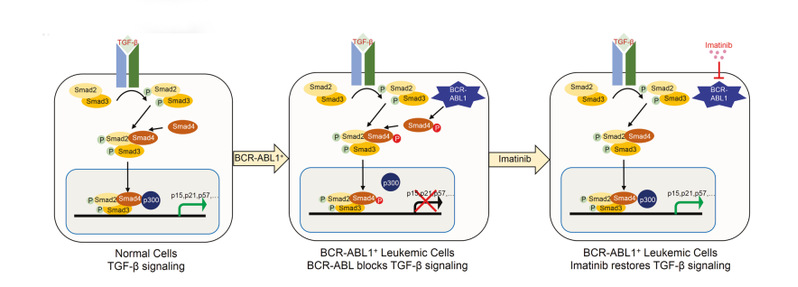
图 1 致癌ABL1阳性肿瘤中TGF-β信号传导中Smad4酪氨酸磷酸化作用的工作模型
从机制上讲,连接区中Smad4在Tyr195、Tyr301和Tyr322的磷酸化干扰其与转录共激活物p300/CBP的结合,从而阻断Smad4激活细胞周期蛋白依赖性激酶(CDK)抑制剂表达和诱导细胞周期停滞的能力。相反,伊马替尼对BCR-ABL1激酶的抑制阻止了Smad4酪氨酸磷酸化,并使CML细胞对TGF-β诱导的抗增殖和促凋亡反应重新敏感。此外,磷酸化位点突变的Smad4的Y195F/Y301F/Y322F突变体在Smad4-null CML细胞中的表达增强了对TGF-β的抗增殖反应,而模仿Y195E/Y301E/Y322E的磷酸化突变体干扰了TGF-β信号传导并增强了CML细胞的体内生长。这些发现证明了BCR-ABL1酪氨酸激酶在CML中抑制TGF-β信号传导的直接作用,并解释了伊马替尼靶向治疗如何恢复有益的TGF-β抗生长反应。
3. HERC3 promotes YAP/TAZ stability and tumorigenesis independently of its ubiquitin ligase activity
(EMBO J. 2023 Jan 4: e111549)
细胞信号转导控制着细胞结构与功能、组织分化与器官发育以及个体生理功能的每一个环节。细胞乃至机体感应和适应环境变化是通过调节细胞信号转导网络来完成的,而细胞信号转导的失常在细胞生长失调和肿瘤发生发展中扮演了重要角色。Hippo信号通路中的转录共激活因子YAP/TAZ的过度激活在肿瘤发生中起着关键作用。在人类恶性肿瘤中也观察到了与Hippo信号无关的YAP/TAZ的异常激活,但其机制在很大程度上仍然不清晰。
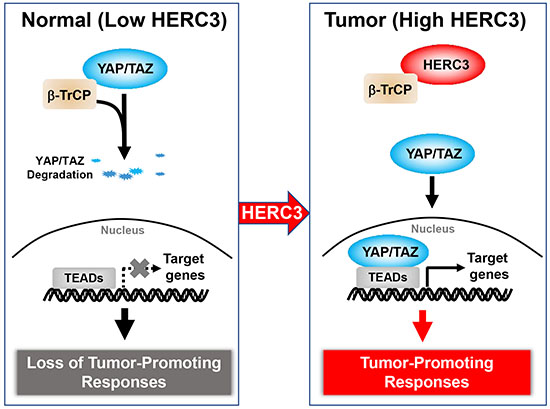
图 1 HERC3促进YAP/TAZ蛋白稳定和肿瘤发生的作用及分子机制工作模型
本研究首次发现YAP/TAZ蛋白的稳定和激活可以被泛素连接酶HERC3所调控,而且不依赖于该泛素连接酶的催化活性。HERC3的减少导致了YAP/TAZ在蛋白水平的降低和下游靶基因表达的减弱。机制研究表明,HERC3可与泛素连接酶β-TrCP的底物识别亚基结合,随后阻断了β-TrCP对底物YAP/TAZ的招募,并拮抗了SCFβ-TrCP介导的YAP/TAZ蛋白泛素化以及被蛋白酶体降解。进一步实验发现,HERC3的缺失抑制了乳腺癌细胞的增殖和转移,该现象可被YAP/TAZ激活突变所逆转,证实HERC3通过增强YAP/TAZ功能促进乳腺肿瘤的发生发展。此外,HERC3在乳腺癌病人样本中高表达,其较高的表达与YAP/TAZ蛋白水平呈正相关,而与患者的生存率呈负相关。上述研究既揭示了调控YAP/TAZ的新机制,也阐明了HERC3的新作用机制。